36. Doublet detection#
Key takeaways
Heterotypic doublets in ADT data can be identified and removed using mutually exclusive cell type markers (e.g., CD3, CD19, CD14), with cells expressing both markers likely representing doublets.
Environment setup
Install conda:
Before creating the environment, ensure that conda is installed on your system.
Save the yml content:
Copy the content from the yml tab into a file named
environment.yml.
Create the environment:
Open a terminal or command prompt.
Run the following command:
conda env create -f environment.yml
Activate the environment:
After the environment is created, activate it using:
conda activate <environment_name>
Replace
<environment_name>with the name specified in theenvironment.ymlfile. In the yml file it will look like this:name: <environment_name>
Verify the installation:
Check that the environment was created successfully by running:
conda env list
name: surface-protein
channels:
- conda-forge
dependencies:
- python=3.13
- scanpy=1.12
- muon=0.1.7
- python-igraph=1.0.0
- ipykernel=7.2.0
- pip==26.0.1
- pip:
- lamindb==2.3.1
- harmonypy==0.0.9
Get data and notebooks
This book uses lamindb to store, share, and load datasets and notebooks using the theislab/sc-best-practices instance. We acknowledge free hosting from Lamin Labs.
Install lamindb
Install the lamindb Python package:
pip install lamindb
Optionally create a lamin account
Sign up and log in following the instructions
Verify your setup
Run the
lamin connectcommand:
import lamindb as ln ln.Artifact.connect("theislab/sc-best-practices").df()
You should now see up to 100 of the stored datasets.
Accessing datasets (Artifacts)
Search for the datasets on the Artifacts page
Load an Artifact and the corresponding object:
import lamindb as ln af = ln.Artifact.connect("theislab/sc-best-practices").get(key="key_of_dataset", is_latest=True) obj = af.load()
The object is now accessible in memory and is ready for analysis. Adapt the
lamindb.Artifact.connect("theislab/sc-best-practices").get("SOMEIDXXXX")suffix to get respective versions.Accessing notebooks (Transforms)
Search for the notebook on the Transforms page
Load the notebook:
lamin load <notebook url>
which will download the notebook to the current working directory. Analogously to
Artifacts, you can adapt the suffix ID to get older versions.
36.1. Motivation#
In the Quality Control chapter, we removed cells that potentially reflect doublets based only on their high count content. We also filtered cells based on sample-wise distribution. Now, we will focus on heterotypic doublets, that is, doublets that contain cells from different cell types. With ADT data, we can detect them using cell type specific surface markers[Sun et al., 2021].
36.2. Environment setup#
import warnings
import muon as mu
import scanpy as sc
warnings.filterwarnings("ignore")
mu.set_options(pull_on_update=False)
sc.settings.verbosity = 0
sc.set_figure_params(
dpi=80,
facecolor="white",
frameon=False,
)
import lamindb as ln
ln.track()
→ loaded Transform('xo1WtNUmrJCK0003', key='doublet_detection.ipynb'), re-started Run('hDXP6BIj1dnyLVwy') at 2026-04-10 17:27:34 UTC
→ notebook imports: lamindb-core==2.3.1 muon==0.1.7 scanpy==1.12
• recommendation: to identify the notebook across renames, pass the uid: ln.track("xo1WtNUmrJCK")
36.3. Loading the data#
We load the MuData object we saved at the end of the previous chapter Normalization:
af = ln.Artifact.connect("theislab/sc-best-practices").get(
key="surface-protein/cite_normalization.h5mu", is_latest=True
)
mdata = af.load()
mdata
MuData object with n_obs × n_vars = 118563 × 36741
var: 'gene_ids', 'feature_types'
2 modalities
rna: 118563 x 36601
obs: 'donor', 'batch'
var: 'gene_ids', 'feature_types'
prot: 118563 x 140
obs: 'donor', 'batch', 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'n_counts', 'outliers'
var: 'gene_ids', 'feature_types', 'n_cells_by_counts', 'mean_counts', 'log1p_mean_counts', 'pct_dropout_by_counts', 'total_counts', 'log1p_total_counts'
layers: 'counts'36.4. Doublets detected with cell type markers#
We are now going to look at cell type markers that are mutually exclusive. Some examples are CD3 (T cell marker) vs CD19 (B cell marker) to identify T/B cells doublets. As cells expressing both specific B and T cell markers do not exist under physiological conditions, those droplets contain T/B cell doublets.
The same is true for cells expressing both T cell (CD3) and monocyte (CD14) markers.
sc.pl.scatter(mdata["prot"], x="CD3", y="CD19-1", color="log1p_total_counts")
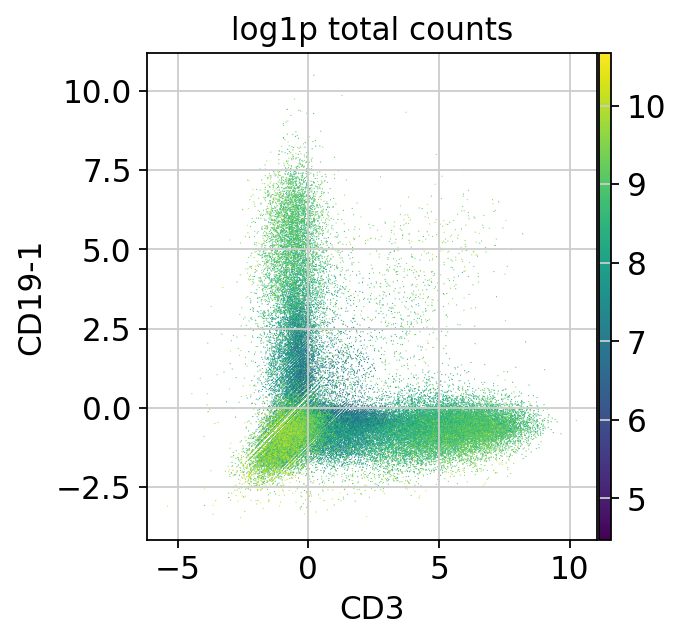
In this plot, we can see a large number of cells not expressing T or B cell markers in the lower left, cells expressing only one marker in the upper left and lower right as well as some cells expressing both markers (upper right).
The cells expressing both markers are doublets and can be removed.
We can also use CD3 and CD14 to detect T/monocyte doublets.
sc.pl.scatter(mdata["prot"], x="CD3", y="CD14-1", color="log1p_total_counts")
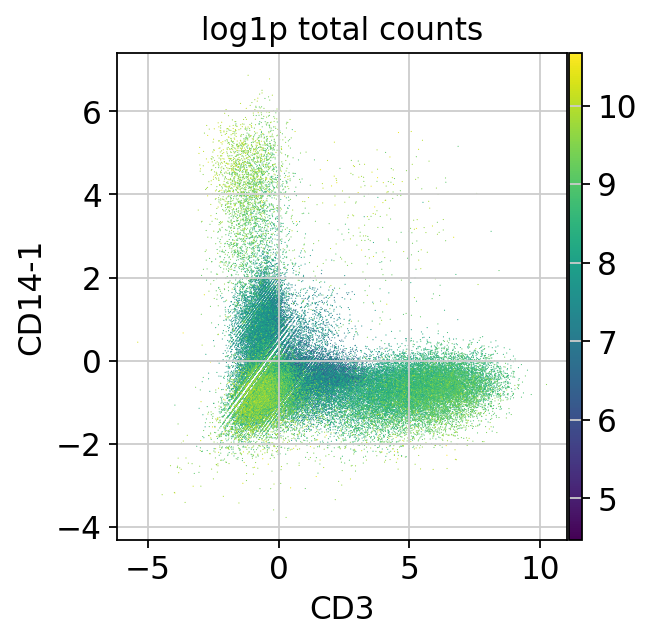
It looks like cells that have an expression level above 2.5 in both markers are doublets. We use an expression level above 2.5 in both markers to flag doublets.
genes2filter = ["CD3", "CD19-1", "CD14-1"]
temp = mdata["prot"][:, genes2filter].X.T.tolist()
mdata["prot"].obs["doublets_markers"] = [
(temp[0][i] > 2.5 and temp[1][i] > 2.5) or (temp[0][i] > 2.5 and temp[2][i] > 2.5)
for i in range(mdata.shape[0])
]
mdata["prot"].obs["doublets_markers"] = (
mdata["prot"].obs["doublets_markers"].astype(str)
)
Doublets usually have a higher count due to the presence of increased counts from more than one cell. We can see this effect in the cells classified as doublets using our markers:
sc.pl.violin(mdata["prot"], keys="log1p_total_counts", groupby="doublets_markers")
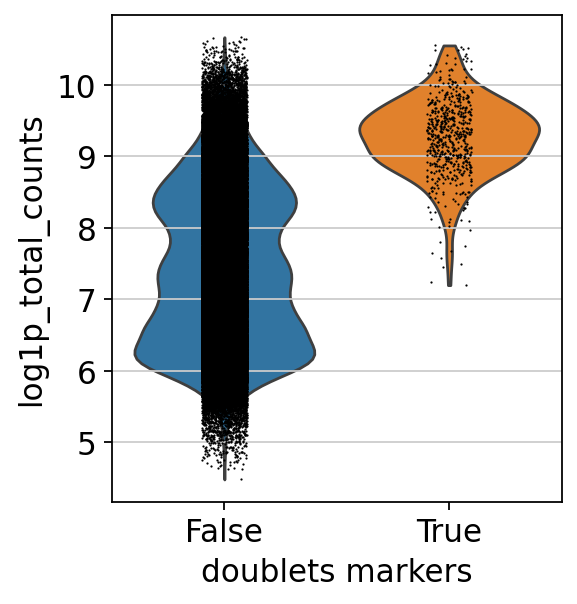
We leave out cells expressing both markers.
mdata = mdata[mdata["prot"].obs["doublets_markers"] == "False"].copy()
mdata
MuData object with n_obs × n_vars = 117951 × 36741
var: 'gene_ids', 'feature_types'
2 modalities
rna: 117951 x 36601
obs: 'donor', 'batch'
var: 'gene_ids', 'feature_types'
prot: 117951 x 140
obs: 'donor', 'batch', 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'n_counts', 'outliers', 'doublets_markers'
var: 'gene_ids', 'feature_types', 'n_cells_by_counts', 'mean_counts', 'log1p_mean_counts', 'pct_dropout_by_counts', 'total_counts', 'log1p_total_counts'
uns: 'doublets_markers_colors'
layers: 'counts'We removed 612 doublets from the data.
In this chapter, we removed doublets using the ADT data by removing cells that highly expressed two mutually exclusive markers. Another option to remove doublets would be utilizing methods that detect doublets based on the scRNA-seq data. For those methods, we refer to the Doublet Detection chapter of scRNA-seq preprocessing and visualization.
af_doublet_detection = ln.Artifact.from_mudata(
mdata,
key="surface-protein/cite_doublet_detection.h5mu",
description="CITE-seq data after doublet detection",
)
af_doublet_detection.save()
ln.finish()
36.5. References#
Bo Sun, Emmanuel Bugarin-Estrada, Lauren Elizabeth Overend, Catherine Elizabeth Walker, Felicia Anna Tucci, and Rachael Jennifer Mary Bashford-Rogers. Double-jeopardy: scrna-seq doublet/multiplet detection using multi-omic profiling. Cell Reports Methods, 1(1):100008, May 2021. doi:10.1016/j.crmeth.2021.100008.
36.6. Contributors#
We gratefully acknowledge the contributions of:
36.6.2. Reviewers#
Lukas Heumos
Anna Schaar