35. Normalization#
Key takeaways
ADT data in CITE-seq requires normalization methods like CLR or dsb to remove noise, with dsb removing ambient and technical noise by using information from empty droplets and isotype controls.
Environment setup
Install conda:
Before creating the environment, ensure that conda is installed on your system.
Save the yml content:
Copy the content from the yml tab into a file named
environment.yml.
Create the environment:
Open a terminal or command prompt.
Run the following command:
conda env create -f environment.yml
Activate the environment:
After the environment is created, activate it using:
conda activate <environment_name>
Replace
<environment_name>with the name specified in theenvironment.ymlfile. In the yml file it will look like this:name: <environment_name>
Verify the installation:
Check that the environment was created successfully by running:
conda env list
name: surface-protein
channels:
- conda-forge
dependencies:
- python=3.13
- scanpy=1.12
- muon=0.1.7
- python-igraph=1.0.0
- ipykernel=7.2.0
- pip==26.0.1
- pip:
- lamindb==2.3.1
- harmonypy==0.0.9
Get data and notebooks
This book uses lamindb to store, share, and load datasets and notebooks using the theislab/sc-best-practices instance. We acknowledge free hosting from Lamin Labs.
Install lamindb
Install the lamindb Python package:
pip install lamindb
Optionally create a lamin account
Sign up and log in following the instructions
Verify your setup
Run the
lamin connectcommand:
import lamindb as ln ln.Artifact.connect("theislab/sc-best-practices").df()
You should now see up to 100 of the stored datasets.
Accessing datasets (Artifacts)
Search for the datasets on the Artifacts page
Load an Artifact and the corresponding object:
import lamindb as ln af = ln.Artifact.connect("theislab/sc-best-practices").get(key="key_of_dataset", is_latest=True) obj = af.load()
The object is now accessible in memory and is ready for analysis. Adapt the
lamindb.Artifact.connect("theislab/sc-best-practices").get("SOMEIDXXXX")suffix to get respective versions.Accessing notebooks (Transforms)
Search for the notebook on the Transforms page
Load the notebook:
lamin load <notebook url>
which will download the notebook to the current working directory. Analogously to
Artifacts, you can adapt the suffix ID to get older versions.
35.1. Motivation#
Similarly to scRNA-seq data, ADT data also contains noise that needs to be removed. The main sources of noise are 1) sequencing depth differences between cells, 2) unspecific binding of antibodies, 3) ambient noise. We aim to remove these sources of noise with normalization techniques.
Contrary to the negative binomial distribution of UMI counts, ADT data is less sparse with a negative peak for non-specific antibody binding and a positive peak resembling enrichment of specific cell surface proteins[Zheng et al., 2022]. The capture efficiency varies from cell to cell due to differences in biophysical properties. Since CITE-seq experiments enrich for a priori selected features, compositional biases are more severe.
Analogously to scRNA-seq data, many approaches to normalization exist. We cover the two most widely used methods.
The traditional way to normalize ADT data, which we cover at the end of this normalization notebook, is using Centered Log-Ratio (CLR) transformation [Stoeckius et al., 2017]. Nevertheless, a new low-level normalization method specifically tailored to dealing with the challenges this modality poses now exists: dsb (denoised and scaled by background) [Mulè et al., 2022]. dsb normalization removes two kinds of noise:
First, dsb normalization uses empty droplets to estimate and to remove ambient noise. This ambient noise arises from free-floating proteins in the solution that are not actually part of a cell. These proteins are most often the result of cell debris from broken cells and are said to be part of the “ambient”. By measuring ADT counts in empty droplets, dsb models the ambient protein levels for each antibody and subtracts this background from droplets containing cells.
Secondly, dsb normalization uses isotype controls to remove noise from unspecific binding. Isotype controls are antibodies that unspecifically bind to many proteins. This information is then used to correct noise arising from unspecific interactions.
35.2. Environment setup#
import warnings
import matplotlib.pyplot as plt
import muon as mu
import pandas as pd
import scanpy as sc
import seaborn as sns
warnings.filterwarnings("ignore")
mu.set_options(pull_on_update=False)
sc.settings.verbosity = 0
sc.set_figure_params(dpi=80, facecolor="white", frameon=False)
import lamindb as ln
ln.track()
→ connected lamindb: theislab/sc-best-practices
→ loaded Transform('Z95qTilYUzGh0002', key='normalization.ipynb'), re-started Run('Ygjmj9z9STgZ7fVF') at 2026-04-10 16:46:01 UTC
→ notebook imports: lamindb==2.2.1 matplotlib==3.10.3 muon==0.1.6 pandas==2.3.1 scanpy==1.11.3 seaborn==0.13.2
• recommendation: to identify the notebook across renames, pass the uid: ln.track("Z95qTilYUzGh")
35.3. Loading the data#
We load the MuData object we saved at the end of the previous chapter Quality Control:
af = ln.Artifact.connect("theislab/sc-best-practices").get(
key="surface-protein/cite_quality_control.h5mu",
is_latest=True,
)
mdata = af.load()
mdata
MuData object with n_obs × n_vars = 118563 × 36741
var: 'gene_ids', 'feature_types'
2 modalities
rna: 118563 x 36601
obs: 'donor', 'batch'
var: 'gene_ids', 'feature_types'
prot: 118563 x 140
obs: 'donor', 'batch', 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'n_counts', 'outliers'
var: 'gene_ids', 'feature_types', 'n_cells_by_counts', 'mean_counts', 'log1p_mean_counts', 'pct_dropout_by_counts', 'total_counts', 'log1p_total_counts'Next, we also load the unfiltered data containing empty droplets for dsb normalization.
mdata_raw contains the unfiltered MuData object with all droplets and mdata contains barcodes that passed the cellranger filtering and our quality control.
While the raw object contains over 24 million droplets, the filtered object only contains 118,563.
af_raw = ln.Artifact.connect("theislab/sc-best-practices").get(
key="surface-protein/cite_raw.h5mu",
is_latest=True,
)
mdata_raw = af_raw.load()
mdata_raw
MuData object with n_obs × n_vars = 24807643 × 36741
var: 'gene_ids', 'feature_types'
2 modalities
rna: 24807643 x 36601
obs: 'donor', 'batch'
var: 'gene_ids', 'feature_types'
prot: 24807643 x 140
obs: 'donor', 'batch'
var: 'gene_ids', 'feature_types'35.4. dsb normalization#
dsb normalization [Mulè et al., 2022] is quite effective in removing noise from ADT data. However, dsb normalization requires two additional sources of data: 1) empty droplets (in this tutorial they are part of the “cite_raw.h5mu” dataset) and 2) isotype controls. If you have neither of these two additional sources of data, you cannot use dsb normalization; in this case we recommend using CLR normalization, which we outline at the end of this normalization section. If you have only one of these additional sources of data, you can use only one of the steps of dsb normalization (not described in this tutorial). If you have both additional sources of data, you can use dsb normalization as outlined in this tutorial.
Isotype controls are antibodies that bind to the cells present in this study non-specifically, meaning you would not expect a significant abundance difference between the cells. Thus, we can use the values of the isotype controls to normalize technical differences.
Let’s first take a look at the raw RNA count distribution in order to tell what droplets are empty droplets and what droplets do contain cells:
sc.pp.calculate_qc_metrics(mdata_raw["rna"], inplace=True, percent_top=None)
sns.displot(
mdata_raw["rna"]
.obs.sample(frac=0.01)
.query("total_counts<100000 and total_counts>10"),
x="total_counts",
log_scale=True,
hue="donor",
multiple="stack",
)
<seaborn.axisgrid.FacetGrid at 0x7fb4b6ff1340>
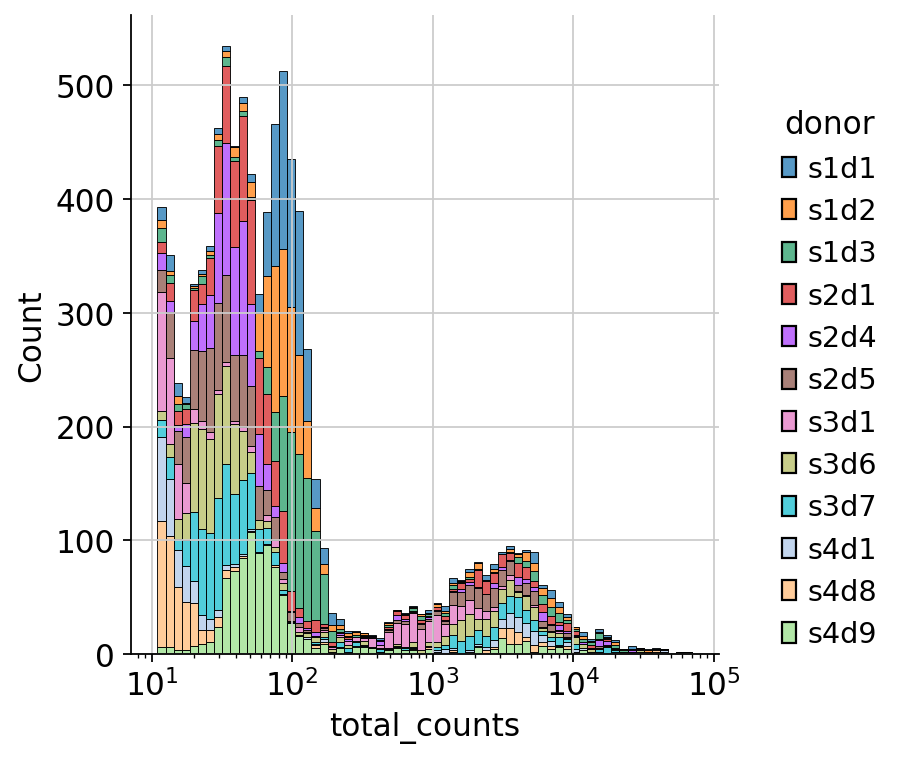
The first large peak between 10**1 and 10**2.1 is composed of droplets that don’t contain cells. We can use these cells for dsb normalization. In this case, the pre-processing software (often CellRanger) already differentiated droplets that contain cells and sorted them into the filtered data, here represented by the “mdata” object. Instead, the unfiltered data, here represented by the “mdata_raw” object, contains all droplets, both with and without cells. dsb normalization will use droplets without cells in order to estimate ambient noise and then remove it.
Additionally, we also now explicitly list the isotype controls that dsb normalization will use to remove noise from unspecific binding.
isotype_controls = ["Mouse-IgG1", "Mouse-IgG2a", "Mouse-IgG2b", "Rat-IgG2b"]
mdata["prot"].layers["counts"] = mdata[
"prot"
].X.copy() # saving the raw data in a layer.
We now call the normalization function muon.prot.pp.dsb() with the filtered and raw mudata object as well as the names of the isotype controls.
mu.prot.pp.dsb(mdata, mdata_raw, isotype_controls=isotype_controls, random_state=0)
Let’s have a look at counts before denoising and normalization.
pd.Series(mdata["prot"].layers["counts"][:100, :100].toarray().flatten()).value_counts()
1.0 1090
0.0 1045
2.0 918
3.0 691
4.0 581
...
457.0 1
132.0 1
633.0 1
374.0 1
763.0 1
Name: count, Length: 524, dtype: int64
See how after denoising and normalization the range changed.
pd.Series(mdata["prot"].X[:100, :100].flatten()).value_counts()
2.499493 1
1.607696 1
-0.490390 1
0.293146 1
-0.099488 1
..
-0.222091 1
-0.208290 1
2.531299 1
0.393625 1
-1.026347 1
Name: count, Length: 10000, dtype: int64
Now let’s take a look at how total protein counts of each cell changed before and after dsb normalization:
sns.histplot(mdata["prot"].layers["counts"].sum(axis=1), bins=50)
plt.title("Total counts per cell (raw)")
plt.xlim(0, 20000)
(0.0, 20000.0)
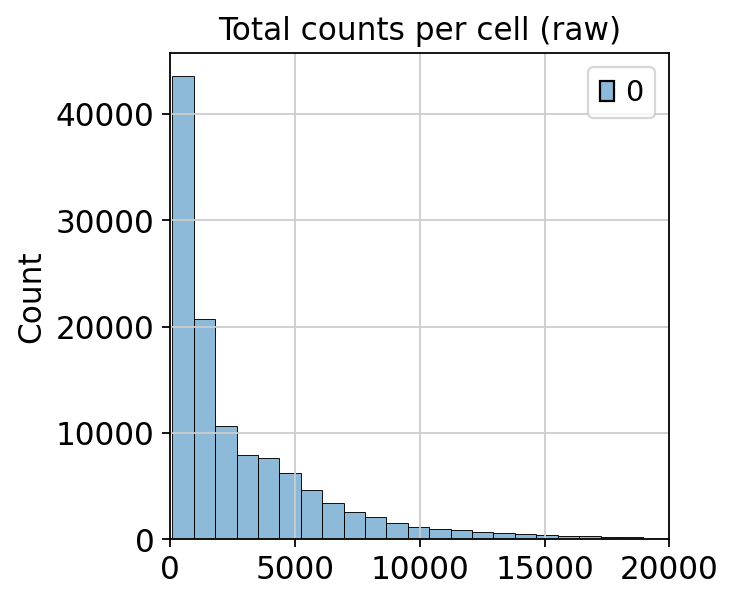
Before normalization, many cells have quite low total protein counts, while some cells have very high total protein counts. These very large differences in total protein counts are largely driven by ambient noise and unspecific binding.
sns.histplot(mdata["prot"].X.sum(axis=1), bins=50)
plt.title("Total counts per cell (dsb normalized)")
plt.xlim(-200, 300)
(-200.0, 300.0)
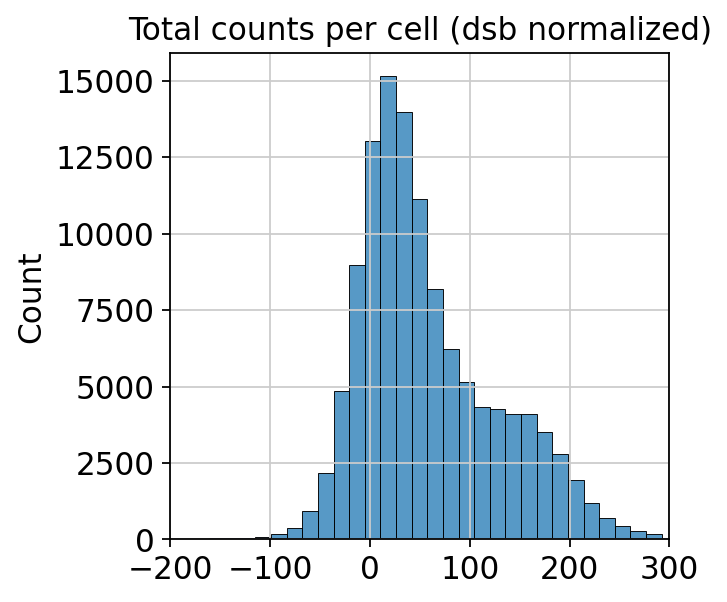
After normalization, the differences in total protein counts are much smaller and (probably) biology-driven rather than noise-driven.
35.5. Centered Log-Ratio normalization#
If you don’t have the unfiltered data and/or the isotype controls available, you can also normalize the ADT data with muon.prot.pp.clr(), implementing Centered Log-Ratio normalization.
CLR normalization is the traditional way of normalizing ADT data, suggested in the original CITE-seq publication [Stoeckius et al., 2017].
CLR normalization accounts for differences in sequencing depth across cells, which can otherwise dominate downstream analyses. Cells with higher total ADT counts may appear artificially distinct, causing biologically similar cells to separate during clustering. CLR normalization rescales protein expression within each cell, making total protein counts more similar in all cells. Specifically, it divides each protein count by the geometric mean of all protein counts in that cell and applies a log transformation.
mdata_clr_normalize = mdata.copy()
mdata_clr_normalize["prot"].X = mdata_clr_normalize["prot"].layers["counts"].copy()
Now we apply CLR normalization by using the function muon.prot.pp.clr():
mu.prot.pp.clr(mdata_clr_normalize["prot"])
We compare the counts before and after normalization:
pd.Series(mdata["prot"].layers["counts"][:100, :100].toarray().flatten()).value_counts()
1.0 1090
0.0 1045
2.0 918
3.0 691
4.0 581
...
457.0 1
132.0 1
633.0 1
374.0 1
763.0 1
Name: count, Length: 524, dtype: int64
pd.Series(mdata_clr_normalize["prot"].X[:100, :100].toarray().flatten()).value_counts()
0.000000 1045
0.416372 33
0.366095 32
0.390344 30
0.429927 30
...
1.092080 1
1.682527 1
0.879271 1
3.436838 1
1.787327 1
Name: count, Length: 3593, dtype: int64
And we plot the distribution of total protein counts per cell:
sns.histplot(mdata_clr_normalize["prot"].X.sum(axis=1), bins=50)
plt.title("Total counts per cell (CLR normalized)")
plt.xlim(0, 400)
(0.0, 400.0)
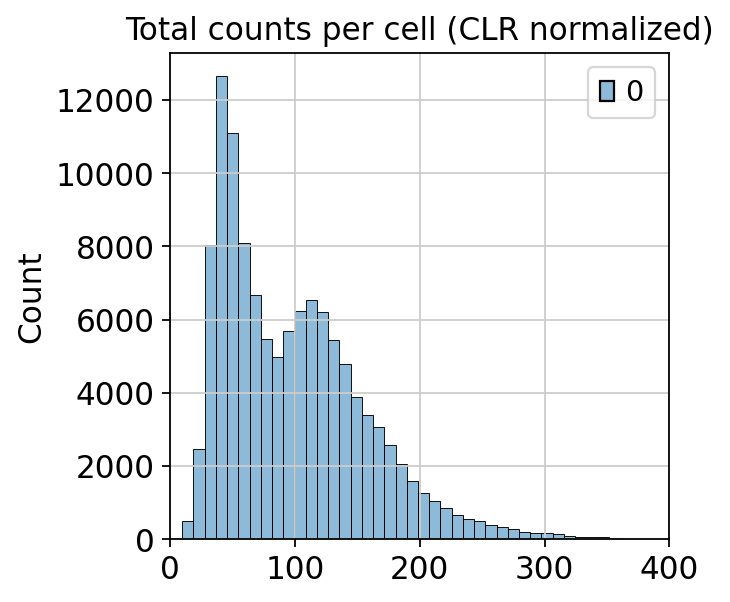
CLR normalization successfully reduced the difference in total protein counts. The range of total protein counts goes from 0 to 350, which is reasonable. If we compare this range with the range of differences in total protein counts in the raw data, which went from 0 to 20000, CLR normalization alleviated some of those differences, which were largely driven by experimental noise.
We save our dsb-normalized CITE-seq data, since dsb normalization is our method of choice when isotype controls and empty droplet data is available.
af_normalization = ln.Artifact.from_mudata(
mdata,
key="surface-protein/cite_normalization.h5mu",
description="CITE-seq data after normalization",
)
af_normalization.save()
ln.finish()
35.6. References#
Matthew P. Mulè, Andrew J. Martins, and John S. Tsang. Normalizing and denoising protein expression data from droplet-based single cell profiling. Nature Communications, 13(11):2099, Apr 2022. doi:10.1038/s41467-022-29356-8.
Marlon Stoeckius, Christoph Hafemeister, William Stephenson, Brian Houck-Loomis, Pratip K. Chattopadhyay, Harold Swerdlow, Rahul Satija, and Peter Smibert. Simultaneous epitope and transcriptome measurement in single cells. Nature Methods, 14(9):865–868, Sep 2017. URL: https://doi.org/10.1038/nmeth.4380, doi:10.1038/nmeth.4380.
Ye Zheng, Seong-Hwan Jun, Yuan Tian, Mair Florian, and Raphael Gottardo. Robust normalization and integration of single-cell protein expression across cite-seq datasets. bioRxiv, 2022. URL: https://www.biorxiv.org/content/early/2022/05/01/2022.04.29.489989, arXiv:https://www.biorxiv.org/content/early/2022/05/01/2022.04.29.489989.full.pdf, doi:10.1101/2022.04.29.489989.
35.7. Contributors#
We gratefully acknowledge the contributions of:
35.7.2. Reviewers#
Lukas Heumos
Anna Schaar